Displaying 3961 - 3975 of 7218

Blog
Behind the scenes with researchers from the Baby’s First Years study
Baby’s First Years (BFY), a J-PAL North America-funded study, recently published results showing that monthly cash support impacts infant brain activity. In an interview with J-PAL staff, J-PAL affiliated professor Lisa Gennetian (Duke University) and researcher Kimberly Noble (Teachers College...
Resource
Basic page
J-PAL's response to Covid-19
Resource
Basic page
Frequently Asked Questions (FAQ) Survei Kesejahteraan
Evaluation
The Impact of Transportation and Information Access on Women's Job Search and Employment in Pakistan
Researchers conducted a randomized evaluation to test the impact of increased access to transportation and assistance with interview scheduling on women’s employment.

Event
Evidence-Informed Policymaking in Germany and Beyond
This event will bring together German domestic policymakers, implementers, and actors of the German development cooperation to discuss and learn about evidence use and rigorous impact evaluation. Together we would like to work towards more evidence-informed programming and policymaking in Germany...
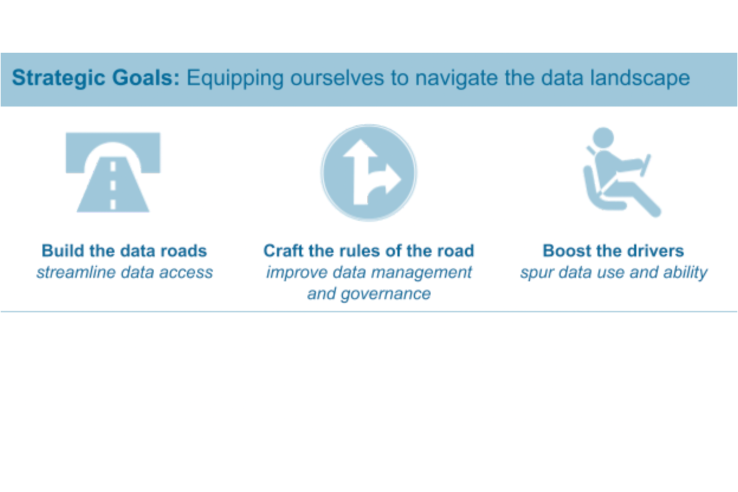
Blog
The road to rigorous evaluation: training partnership builds use of evidence in California
This post shares reflections from Joy Bonaguro, California Statewide Chief Data Officer, and Jason Lally, Deputy Chief Data Officer. In September 2021, CalData and J-PAL North America partnered to provide a training on Designing Rigorous Evaluations of Government Programs to participants from...

Blog
End the pandemic of violence against women
To end violence against women and girls requires effective prevention strategies that target its root causes. Such programs address pervasive inequalities and power differentials—especially gender norms that justify and normalize violence against women and girls.

Update
J-PAL Updates
February 2022 J-PAL North America Newsletter
J-PAL North America's February newsletter features a new evidence review on sectoral employment programs, research results from a study of financial aid take-up, and ways we are leveraging tutoring evidence to inform federal policy conversations.

Policy insight
Improving student learning: Impacts by gender
Most programs to improve student learning have similar impacts on girls and boys. However, policymakers should consider potential different effects by gender while designing programs since, in some cases, program design choices led to different impacts on girls and boys.

Blog
J-PAL South Asia launches ASPIRE with US$6.3 million grant from Veddis Foundation to strengthen the culture of evidence-based policymaking in India
J-PAL South Asia’s efforts to promote policymaking based on scientific evidence and data in India has received a major boost with the launch of the Alliance for Scaling Policy Impact through Research and Evidence (ASPIRE) in partnership with the Veddis Foundation.

Update
J-PAL Updates
Veddis Foundation and J-PAL South Asia Set Up ASPIRE to Accelerate Evidence-Based Policymaking
New Delhi, India: The Abdul Latif Jameel Poverty Action Lab (J-PAL) South Asia and Veddis Foundation announced the launch of the Alliance for Scaling Policy Impact through Research and Evidence (ASPIRE) today. ASPIRE aims to spur the adoption of anti-poverty policies rooted in scientific evidence...

Update
J-PAL Updates
Government of Delhi Partners with J-PAL South Asia to Unlock High-Quality Administrative Data for Boosting Social and Economic Growth in India’s Capital
The Government of Delhi’s Dialogue and Development Commission (DDC) has partnered with the Abdul Latif Jameel Poverty Action Lab (J-PAL) South Asia to systematically leverage high-quality administrative data for implementing policies geared toward stimulating social and economic development in the...
Resource
Basic page
Register for Evidence-Informed Policymaking in Germany and Beyond
Registration is now closed.
Resource
Basic page
Register to attend the launch of the Egypt Impact Lab
The launch of the Egypt Impact Lab was held on Thursday, March 17, 10:00 am to 1:30 pm EET, at the Moataz Al Alfi Hall, AUC New Cairo.

Event
Launching the Egypt Impact Lab to strengthen evidence-informed policy and improve development outcomes for Egyptians
The Egyptian Ministry of Planning and Economic Development and the Abdul Latif Jameel Poverty Action Lab Middle East and North Africa at the American University in Cairo are proud to announce the launch of the Egypt Impact Lab. Learn about its partners' top priorities and the course for conducting...