
Resource
Layout Page

For more information on statistical power and how to perform power calculations, see the Power Calculations resource; for additional technical background and sample code, see the Quick Guide to Power Calculations; for practical tips on conducting power calculations at the start of a project and additional intuition behind the components of a well-powered study, see Six Rules of Thumb for Determining Sample Size and Statistical Power.
This resource is intended for researchers in the social sciences who are considering publishing their randomized evaluation in a medical journal. Publishing in a medical journal allows researchers to share their study with researchers and practitioners in different disciplines and expand the reach of their findings to those outside of their immediate professional network. This resource highlights journal guidelines and requirements that may be unfamiliar to those used to publishing in the social sciences. Most medical journals are members of the International Committee of Medical Journal Editors (ICMJE) and follow its recommendations and requirements for publishing.1 Some of the requirements for publishing necessitate action before a study has even started. While most of the content in this checklist is relevant across all medical journals, certain elements may differ from journal to journal. Researchers are encouraged to consult requirements listed in this resource in conjunction with the requirements of their specific journal(s) of interest, as well as requirements from funders and universities that could influence the publication process.
The ICMJE defines a clinical trial as, “any research study that prospectively assigns human participants or groups of humans to one or more health-related interventions to evaluate the effects on health outcomes.” If a randomized evaluation meets the definition of a clinical trial, researchers must register it in an ICMJE compliant registry before the first participant is enrolled. If researchers fail to meet this condition, they risk having their paper rejected by medical journals. Note that registration on the AEA’s RCT Registry (supported by J-PAL) does not meet this requirement.
Not every randomized evaluation that researchers submit to a medical journal will meet the ICMJE definition of a clinical trial and require registration. However, if it is unclear whether a study meets the definition of a clinical trial, follow the registration guidelines for clinical trials as a cautionary measure. This way if a journal editor feels that a study is a clinical trial, researchers will have met the registration requirement.
Some scenarios where registration prior to enrollment may be unnecessary (although different journals may reach different conclusions) include the following:
More information on trial registration, including J-PAL requirements, can be found in this resource on trial registration. Information on labeling a journal submission that is not a clinical trial is discussed below.
A trial protocol outlines the objectives, methodology, study procedures, statistical analysis plan (SAP), and ethical considerations of a study. This is the same protocol that researchers must submit to an institutional review board (IRB) prior to beginning human subjects research. Medical journals require the trial protocol to be submitted at the time of manuscript submission. Because IRBs may vary in what information they request, researchers should use the SPIRIT reporting guidelines when preparing protocols to ensure completeness. An example is the trial protocol (with an integrated SAP) for Health Care Hotspotting.
Researchers should plan to submit the original approved protocol along with all amendments and their rationale. Amendments should always be reviewed and approved by an IRB prior to taking effect. Significant changes to the original protocol should be described in any future publications. The trial protocol is typically included with a publication as an online supplement.
A statistical analysis plan (also known as a pre-analysis plan) provides detailed information on how data will be coded and analyzed. A SAP can be integrated into a trial protocol or may exist as a separate document. Most medical journals require a SAP to be submitted along with the manuscript.
Unlike trial registration, journals differ on when a SAP must be completed. However, for some journals, completing a SAP prior to the launch of the intervention, or at least prior to receiving data and beginning analysis, can increase the credibility of findings and/or allow researchers greater freedom in using causal language when interpreting results. Defining analysis plans in advance also reduces concerns about specification searching (p-hacking), particularly when there is researcher discretion about what outcomes to consider and when and how they are measured. Most medical journals will require researchers to indicate which analyses are prespecified and which are post hoc. As with the trial protocol, the final SAP should be submitted to the journal, with any changes to original plans tracked and justified.
J-PAL does not universally require SAPs, but many individual J-PAL initiatives and funders require that an analysis plan be completed prior to the launch of an evaluation. For example, all J-PAL North America funded projects must upload a SAP to the AEA RCT Registry. See this resource on pre-analysis plans for more detailed information and recommended practices.
Guidelines for the Content of Statistical Analysis Plans in Clinical Trials provides a checklist that researchers can follow to help structure their SAPs. A detailed explanation of each section can be found in Appendix 2 of the article. Some important topics to cover in a SAP include:
Ensure primary and secondary outcomes are prespecified. Defining a single primary outcome aids in interpretation of the overall result of the research, particularly if multiple outcomes yield different results. The primary outcome should be one that the study is well powered to detect, whereas important outcomes that researchers may be less confident observing effects for can be listed as secondary outcomes. For example, in medical research a short term clinical endpoint may be selected as a primary outcome, with mortality selected as a secondary outcome. Include details on how and when outcomes will be measured, their units, and any calculations or transformations made to derive outcomes.
Include statistical models, any plans for covariate adjustment, and additional analysis beyond the intention to treat effect, such as local average treatment effect (LATE), and approaches to noncompliance or partial take-up.
Outline how missing data will be dealt with and reported. Attrition due to imperfect matches in administrative data and survey non-responses are both situations where researchers will need to plan to address missing data. Medical journals often have strong opinions about how to handle missing data and may request sensitivity analysis or multiple imputation even when missingness is low. Defining an approach in advance will reduce concerns about specification searching. The plan may include flexibility depending on the severity of missing data. See the Journal of the American Medical Association's (JAMA) guidelines on missing data and the New England Journal of Medicine's (NEJM) recommendations for handling missing data.
Testing multiple hypotheses increases the probability of false positive results. Therefore, researchers should have a plan to address multiple testing when designing an evaluation. Absent a prespecified approach, journals may treat results for secondary outcomes as only exploratory. The multiple hypothesis testing section of this resource on data analysis has suggestions on statistical corrections when examining multiple outcomes.
Medical journals typically require significant detail on how participants progress through an evaluation. During an evaluation’s implementation stage, researchers should keep track of participants’ study involvement, the mechanics of randomization, and fidelity to the study protocol.
Researchers should track the flow of participants as they move through each stage of the trial and should report this in their paper. The Consolidated Standards of Reporting Trials’ (CONSORT) Statement, a minimum set of recommendations for reporting RCTs, recommends using this diagram (see Figure 2) to display participant flow. All trials published in medical journals include this CONSORT flow diagram, and one should always be included as a figure in your paper (typically the first exhibit). See an example of a CONSORT flow diagram below.
At a minimum, researchers should track the number of individuals randomly assigned, the number who actually receive the intervention, and the number analyzed. Information on individuals assessed for eligibility prior to randomization can be included if available. Any additional information on exclusions or attrition (also known as loss to follow-up) should be included as well.
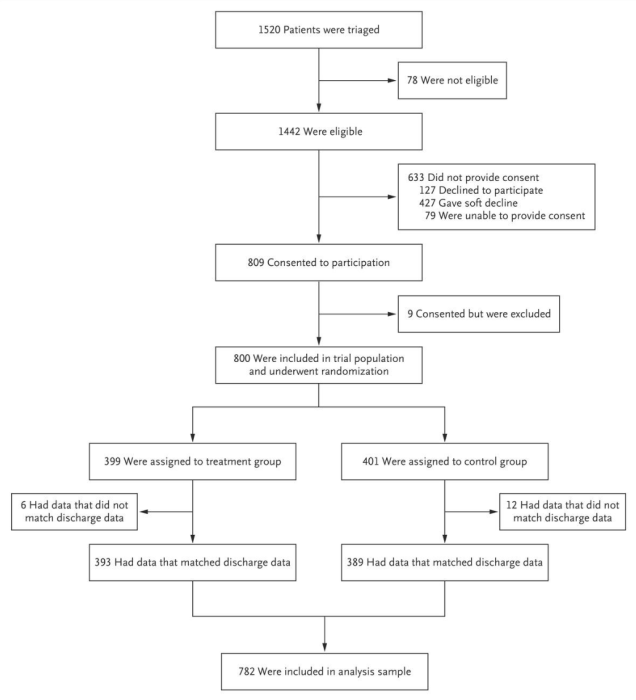
Source: Finkelstein et al. 2020
Researchers should keep track of the randomization process and the mechanics of implementation, as these details must be reported in detail in a manuscript. This typically includes the type of randomization procedure — was randomization stratified, done on the spot or from a list — as well as who generated random assignments, how the information was concealed or protected from tampering, and how information was revealed to participants. Items 8-10 of the CONSORT checklist cover reporting on randomization processes. See Financial Support to Medicaid-Eligible Mothers Increases Caregiving for Preterm Infants for an example of reporting these details in a manuscript.
The CONSORT statement notes that “the simple way to deal with any protocol deviations is to ignore them.” This is consistent with the intention to treat approach to randomized evaluation. However, providing detail is helpful to contextualize and interpret results. See the resource on implementation monitoring and real-time monitoring and response plans for guidance. Researchers approach monitoring implementation and reporting on it in different ways.
Clinical trial reporting in medical journals follows a specific style characterized by consistent structure, short word counts, and a small number of tables and figures. The Consolidated Standards of Reporting Trials’ (CONSORT) Statement provides a set of standardized recommendations, in the form of a checklist and flow diagram, for reporting randomized evaluations in medical journals. Researchers should consult the CONSORT Statement, as well as particular journal requirements, in order to ensure a manuscript is properly formatted and structured. This section highlights elements of analysis and manuscript preparation that may be less familiar to social scientists.
CONSORT guidelines state that researchers should not report statistical tests of balance in participant characteristics, and many medical journals follow this recommendation. This practice differs from a common norm in economics to report balance tests in summary statistics tables. Researchers can find more information on the logic behind this rule in the CONSORT Explanation and Elaboration resource.
Results should be reported for all primary and secondary outcomes, regardless of their statistical significance. Detailed results should be reported for each outcome, typically mean levels for each group, treatment effect, confidence interval, and p-value. Confidence intervals are preferred to standard errors. For binary outcomes, both absolute and relative effects should be reported. Researchers should make clear which analyses are prespecified, particularly in the case of subgroup analyses.
Below is an example of a results table with multiple outcomes from Health Care Hotspotting. Each row highlights one outcome, with mean levels for each group, an unadjusted group difference (from a regression without controls), an adjusted group difference (from a regression with controls), and corresponding confidence intervals. In this case, all analyses were prespecified and p-values were omitted.
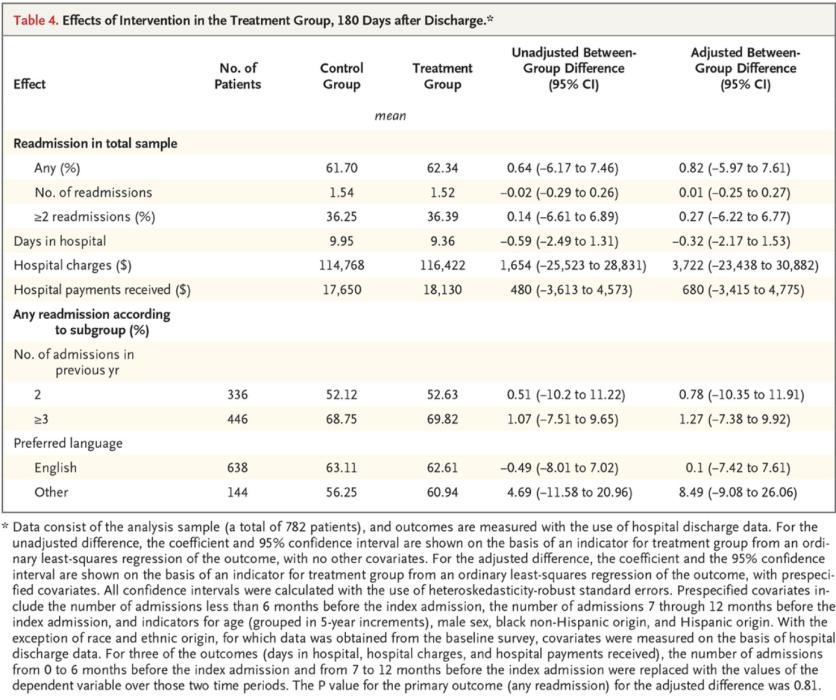
Source: Finkelstein et al. 2020
Journals typically cap the total number of exhibits (tables and figures) at five. Figure 1 should typically be the participant flow diagram. A summary statistics table, typically Table 1, should always describe baseline demographic and clinical characteristics for each group. The remaining figures and tables display the results of the evaluation.
For studies with multiple treatment arms or subgroup analyses, it may be helpful to use an exhibit with multiple panels or a hybrid exhibit that combines a table and a figure to provide more detail. Figure 2 in Effectiveness of Behaviorally Informed Letters on Health Insurance Marketplace Enrollment gives an example of a hybrid exhibit. Note, however, that some journals may not view this as a single exhibit. Additional exhibits may be included in a supplemental appendix.
Before preparing a manuscript for a medical journal, you should consult the specific word and exhibit limits of that journal as well as its requirements on how the manuscript should be structured. Compared to social science publications, reports of clinical trials in medical journals are consistently structured and short (e.g., 3000 words) with binding limits on the number of words and exhibits. Medical journals are generally much more prescriptive about what each section in the paper must accomplish. Medical journals do allow for supplemental appendices where researchers can include details that did not fit in the main body of the paper. Most journals follow CONSORT reporting requirements. Consult the CONSORT checklist as you write and make sure to include all sections. Some journals require submission of the checklist to ensure that all elements have been addressed.
Some key requirements from the CONSORT checklist have been highlighted below:
Include ‘randomized’ in the manuscript title so that databases properly index the study and readers can easily identify the study as a randomized trial.
The abstract should be framed as a mini paper, with clear headings, and include a sufficient summary of the work, including results and interpretation. The abstract is not a preview that raises questions to be answered in the main text. Effort should be focused here because this is the first section that will be reviewed and often the only section that many readers will read. A full list of what to include can be found within Item 1b. of the CONSORT Statement. Trial registration information (registry name, trial ID number) should be listed at the end of the abstract.
Despite the short length of medical journal articles, the methods section must include a high level of detail. Required details include mechanisms of random assignment, power calculations, intake and consent procedures, where IRB approval was granted, statistical analysis methods, and software packages used.
Participant flow and baseline characteristics should be described first, followed by the effects of the intervention. Results are presented in text with similar detail to the tables discussed above (i.e., control and treatment group means, effect sizes, confidence intervals). The results section presents results without interpretation, which is reserved for the discussion section.
The discussion section offers the opportunity to discuss limitations and generalizability, and provide additional interpretation. This section is where researchers can discuss and weigh the sum total of the evidence if results across outcomes are mixed, or draw comparisons to results in other scholarship. This section also offers an opportunity to summarize and conclude, though information provided in other parts of the paper should not be repeated in detail. If a journal allows a separate conclusion section it is typically a single summary paragraph.
Researchers should ensure their paper is properly pitched for a medical or public health audience rather than an economics audience. Consulting the stated aims and scope of a journal can help with framing study results and emphasizing its relevance to the field. Highlighting the clinical rationale and patient care implications of a study can be helpful as medical journal readership is often interested in these factors. If possible, researchers should have someone who has frequently published in medical journals review their manuscript before submission to make sure the manuscript is properly framed for the intended audience.
Submit your manuscript for review and publication
There are some additional considerations to keep in mind at the time of submission:
Journals may require submission of a cover letter along with a manuscript. There are conflicting opinions on whether or not the cover letter is important. One view is that it needs to say nothing more than “Thank you for considering the attached paper.” Another view is that the cover letter is an opportunity to pitch the paper and explain why the findings are important and relevant to the readers of this journal. It can also be used to provide context for information included elsewhere in the submission, such as whether there are preprints of the paper or potential conflicts of interest.
When submitting a paper to a journal, researchers typically select what type of study is being submitted. If the study is labeled as a clinical trial, journals will expect the study to have been registered prior to enrollment. If this was not the case, this may lead the paper to be automatically rejected. If the study is a secondary analysis of a clinical trial, make sure to specify this. Journals may list secondary analyses as a subcategory of clinical trials or may list them as a separate type. A detailed definition of clinical trials and considerations for trial registration are discussed in Section 1 of this resource.
Journals may be reluctant to publish studies or results that are already available online. Distributing a manuscript as a working paper or uploading it to a preprint server may lead journals to de-prioritize publication of a paper. Researchers should confirm the policies at their target journal(s) prior to releasing a working paper. The BMJ, The Lancet, NEJM, and JAMA do not penalize studies that are already available as preprints as long as researchers provide information about the preprint during the submission process. Other journals may accept manuscripts only if they have been significantly revised after being released as a working paper.
Most journals impose embargoes where authors cannot disclose paper acceptance or share results until the work has been published. Most journals allow researchers to present at conferences, but may impose restrictions, such as prohibiting researchers from sharing complete manuscripts, and may request that researchers inform the journal of planned presentations. Embargoes will typically include a post-acceptance window where the media has access to upcoming publications, in order to conduct interviews and prepare articles that can be released once the manuscript is published.
Researchers should consult individual journals to ensure they are adhering to their specific embargo rules. Researchers working with implementing partners should ensure that everyone understands and can abide by the embargo policy, and both parties should work together to develop communication plans to take advantage of media access periods. This resource on communicating with a partner about results has additional details on embargo policies and communication plans. While embargoes may seem restrictive, medical journals tend to publish frequently, so the publishing timeline and embargo period can often move along quickly.
A selection of J-PAL affiliated-studies that have been published in medical journals has been highlighted below.
Acknowledgments: Thanks to Marcella Alsan, Catherine Darrow, Joseph Doyle, Laura Feeney, Amy Finkelstein, Ray Kluender, David Molitor, Hannah Reuter, and Adam Sacarny for their thoughtful contributions. Amanda Buechele copy-edited this document. Creation of this resource was supported by the National Institute On Aging of the National Institutes of Health under Award Number P30AG064190. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
1 The full list of journals that follow ICMJE recommendations can be found on ICMJE.org.
Finkelstein, Amy, Annetta Zhou, Sarah Taubman, and Joseph Doyle. “Health Care Hotspotting — A Randomized, Controlled Trial.” New England Journal of Medicine 382, no. 2 (January 9, 2020): 152–62. https://doi.org/10.1056/NEJMsa1906848.
Finkelstein, Amy, Annetta Zhou, Sarah Taubman, Joseph Doyle, and Jeffrey Brenner. "Health Care Hotspotting: A Randomized Controlled Trial." AEA RCT Registry. (2014) https://doi.org/10.1257/rct.329.
Finkelstein, Amy and Jeffrey Brenner. “Health Care Hotspotting: A Randomized Controlled Trial.” Clinicaltrials.gov. (March 18, 2014). https://clinicaltrials.gov/ct2/show/NCT02090426.
Finkelstein, Amy, Yunan Ji, Neale Mahoney, and Jonathan Skinner. “Mandatory Medicare Bundled Payment Program for Lower Extremity Joint Replacement and Discharge to Institutional Postacute Care: Interim Analysis of the First Year of a 5-Year Randomized Trial.” JAMA 320, no. 9 (September 4, 2018): 892–900. https://doi.org/10.1001/jama.2018.12346.
Finkelstein, Amy, Yunan Ji, Neale Mahoney, and Jonathan Skinner. “The Impact of Medicare Bundled Payments.” Clinicaltrials.gov. (January 23, 2018) https://clinicaltrials.gov/ct2/show/NCT03407885
Gamble, Carrol, Ashma Krishan, Deborah Stocken, Steff Lewis, Edmund Juszczak, Caroline Doré, Paula R. Williamson, et al. “Guidelines for the Content of Statistical Analysis Plans in Clinical Trials.” JAMA 318, no. 23 (December 19, 2017): 2337–43. https://doi.org/10.1001/jama.2017.18556.
Hinman, Rana S., Rachelle Buchbinder, Rebecca L. Craik, Steven Z. George, Chris G. Maher, and Daniel L. Riddle. “Is This a Clinical Trial? And Should It Be Registered?” Physical Therapy 95, no. 6 (June 1, 2015): 810–14. https://doi.org/10.2522/ptj.2015.95.6.810.
J-PAL. “Health Care Hotspotting in the United States.” https://www.povertyactionlab.org/evaluation/health-care-hotspotting-united-states
J-PAL. “The Spillover Effects of a Nationwide Medicare Bundled Payment Reform.” https://www.povertyactionlab.org/evaluation/spillover-effects-nationwide-medicare-bundled-payment-reform
Reif, Julian, David Chan, Damon Jones, Laura Payne, and David Molitor. “Effects of a Workplace Wellness Program on Employee Health, Health Beliefs, and Medical Use: A Randomized Clinical Trial.” JAMA Internal Medicine 180, no. 7 (July 1, 2020): 952–60. https://doi.org/10.1001/jamainternmed.2020.1321.
Song, Zirui, and Katherine Baicker. “Effect of a Workplace Wellness Program on Employee Health and Economic Outcomes: A Randomized Clinical Trial.” JAMA 321, no. 15 (April 16, 2019): 1491–1501. https://doi.org/10.1001/jama.2019.3307.
Ware, James H., David Harrington, David J. Hunter, and Ralph B. D’Agostino. “Missing Data.” New England Journal of Medicine 367, no. 14 (October 4, 2012): 1353–54. https://doi.org/10.1056/NEJMsm1210043.
Yokum, David, Daniel J. Hopkins, Andrew Feher, Elana Safran, and Joshua Peck. “Effectiveness of Behaviorally Informed Letters on Health Insurance Marketplace Enrollment: A Randomized Clinical Trial.” JAMA Health Forum 3, no. 3 (March 4, 2022): e220034. https://doi.org/10.1001/jamahealthforum.2022.0034.



